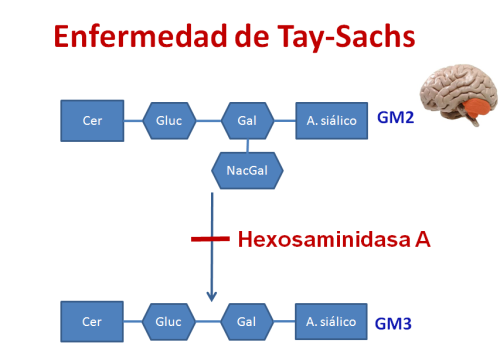
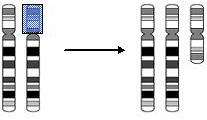
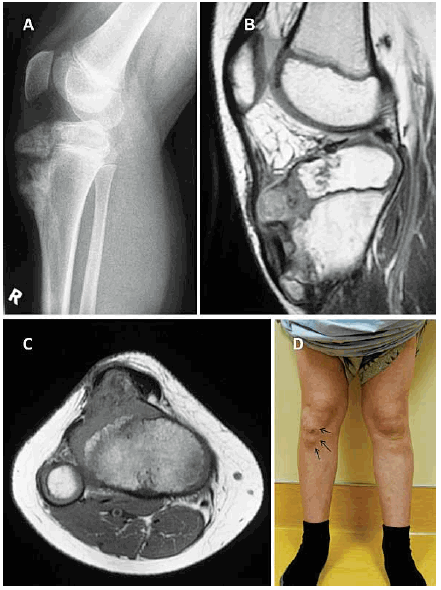
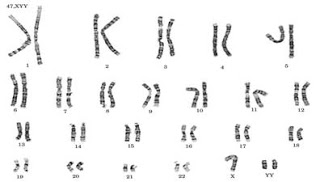
Because who is perfect?
Hola queridos lectores, hoy no vamos a hablar de ninguna enfermedad rara en concreto, vamos a finalizar este blog haciéndolo de todas a la vez. Las enfermedades huérfanas comparten unas impactantes características por las cuales todos debemos tener un gran respecto hacia todos aquellos que las padecen; por lo general son graves y degenerativas, llegan a producir la incapacitación de la persona, con una negativa y alta carga psicosocial, incurables... Y todas ellas no afectan únicamente a los pacientes que las padecen sino a su entorno, a sus familias, a sus amigos.. Por todos estos motivos debemos tener EMPATÍA y pensar que mañana podemos ser nosotros quien nos encontremos en su piel. No permitáis aumentar el número de individuos que tienen desconocimientos acerca de las patologías raras y por tanto las excluyen de la sociedad. Sumaros al bando del conocimiento, de la comprensión, ayudando a estos enfermos a llevar una vida mejor.
Es necesario desarrollar una política nacional de salud pública respecto a las enfermedades raras la cual permita mayor investigación científica acerca de las mismas, de medicamentos, cuidados sociales, hospitalización y tratamiento... y sería éticamente correcto hacer todo esto con fondos públicos destinados para ello. Es indignante que algunas personas con estas patologías no puedan ser atendidos por los sistemas sanitarios públicos, el insuficiente conocimiento de la población en general, de la ciencia en dicho campo, el escabullimiento de las autoridades de la nación competentes, de las farmacéuticas...y que la creación de asociaciones de enfermos y familiares sea su única salida. No permitamos que nadie sea invisible en la sociedad, porque juntos podemos!