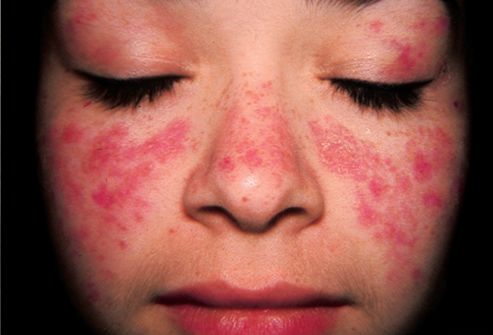
El Síndrome de Bloom o síndrome de rotura cromosómica es una enfermedad rara caracterizada por una inestabilidad genética asociada con un retraso en el crecimiento pre y postnatal, hipersensibilidad a la luz solar que da lugar a una erupción facial rojiza en la piel de las mejillas o en el dorso de las manos, aumento de la sensibilidad a infecciones y predisposición al cáncer.
También pueden presentar telangiectasia, baja estatura (la estatura media en la edad adulta es de 150 cm), voz aguda, caras estrechas y largas con nariz y orejas prominentes, áreas hipo e hiperpigmentadas etc.
Su prevalencia global es desconocida actualmente pero se han descrito más de 100 casos.

Esta enfermedad se debe a mutaciones en el gen BLM que codifica una enzima implicada en la tarea de desenrollar el ADN en procesos importantes tales como la replicación, la transcripción y la reparación de ADN.
Las personas que padecen la enfermedad tienen un enorme aumento en el intercambio entre los fragmentos de cromosomas homólogos o cromátidas hermanas,y, además, aumentos de roturas cromosómicas y reordenamientos en comparación con las personas sanas.
Se trata de una enfermedad de transmisión autosómica recesiva, siendo el riesgo de recurrencia del 25%. Esto quiere decir que ambos padres deben ser portadores para que el niño esté afectado.
El diagnóstico clínico se confirma mediante citogenética y mediante pruebas de genética molecular.
El diagnóstico prenatal puede realizarse mediante una amniocentesis.
El tratamiento es sintomático. Se emplean antibióticos para tratar las infecciones. En caso de que los niveles séricos de inmunoglobulinas sean bajos, los pacientes se beneficiarán de una terapia de substitución de estas. También debe evitarse la exposición al sol.
Además, el objetivo último del tratamiento es corregir quirúrgicamente las deformidades óseas y tratar rápidamente las posibles tumoraciones malignas que aparezcan,
La esperanza de vida no sobrepasa la edad adulta tardía, aproximadamente 50 años de edad.
En esta página tenéis más información!!
Fuentes:
http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=125
http://www.ecured.cu/S%C3%ADndrome_de_Bloom